EMA’s Draft Guideline: Non-Inferiority & Equivalence Comparisons in Clinical Trials
- Sharan Murugan

- 35 minutes ago
- 3 min read
The European Medicines Agency (EMA) has published the Draft Guideline on Non-Inferiority and Equivalence Comparisons in Clinical Trials, a major methodological update replacing earlier guidance documents and modernising expectations for confirmatory clinical trial design.
This new guidance introduces updated statistical concepts, integrates the ICH E9(R1) estimand framework, and clarifies how sponsors should design, analyse, and interpret non-inferiority and equivalence trials across therapeutic areas.
Non-inferiority means showing that a new treatment is not unacceptably worse than an active comparator by more than a pre-specified clinical margin. The intention is to demonstrate that the new treatment retains enough of the comparator’s effect to still be clinically valuable.
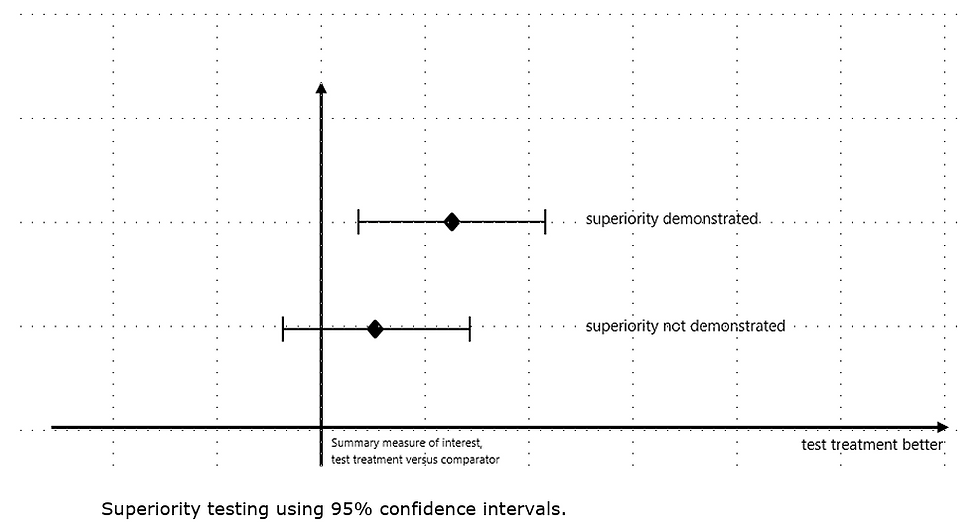
1. Why This New EMA Draft Guideline Matters
The guidance highlights that these designs are significantly more complex than superiority trials.The guideline explains that demonstrating non-inferiority or equivalence requires strong assumptions about assay sensitivity, constancy of effect, and historical comparator performance. It also reminds sponsors that even when the non-inferiority or equivalence hypothesis is statistically met, EMA will still assess the totality of evidence for benefit–risk.
2. Scope of the Guideline
According to the guidance, this guidance applies to confirmatory clinical trials that assess:
Absolute efficacy versus putative placebo
Relative efficacy versus active comparator
Non-inferior safety
Biosimilarity based on clinical efficacy
Therapeutic equivalence
Equivalence in pharmacodynamic measures
It explicitly excludes bioequivalence, PK studies, or CMC quality assessments.
3. Clear Definitions of Trial Objectives
The guidance outlines three primary objectives for non-inferiority comparisons:
• Absolute efficacy
Used when a placebo-controlled trial is unethical; an active comparator stands in for placebo.
• Relative efficacy
The aim is to show the new treatment is not worse than an active comparator by more than the accepted margin.
• Non-inferior safety
Designed to demonstrate that a new therapy does not increase risk beyond a clinically unacceptable amount.
For equivalence trials, the guideline defines the objective as showing that two treatments are “similar enough” to be deemed therapeutically equivalent, often in biosimilar development.
4. Statistical Foundations Explained
The guidance includes clear graphical explanations showing how superiority, non-inferiority, and equivalence are demonstrated using two-sided 95% confidence intervals.The guideline states that non-inferiority requires the CI to lie above –Δ, while equivalence requires it to fall between –Δ and +Δ, ensuring control of type-I error.
5. Assay Sensitivity & Constancy Assumption
The guideline emphasises that assay sensitivity is essential for valid non-inferiority and equivalence results.The guidance notes that poor conduct, inconsistent patient selection, or protocol deviations can artificially reduce differences between treatments.When no placebo arm exists, applicants must justify the constancy assumption, showing that historical comparator performance is still relevant to today’s trial setting.
6. Estimands Must Align with Non-Inferiority and Equivalence Objectives
The guideline incorporates ICH E9(R1) and requires that the primary estimand reflects both the scientific and regulatory intent behind the non-inferiority or equivalence comparison.The guidance stresses that single estimands often cannot answer all regulatory questions, making supplementary estimands necessary. It highlights that estimands used for superiority may not be appropriate for non-inferiority settings.
7. Margin Selection: One of the Most Critical Elements
For absolute efficacy
The guideline explains that the non-inferiority margin should preserve a meaningful fraction of the active comparator’s effect over placebo. It describes two approaches:
Fixed-margin approach
Synthesis approach
Both rely heavily on historical evidence and constancy.The guideline provides detailed formulas and comparisons.
For relative efficacy or equivalence
Margins must be clinically justified, not simply based on effect retention.The guidance states that margins cannot be inflated just to make sample sizes feasible, and they must reflect differences that are acceptable to patients and clinicians.
8. Handling Missing Data & Intercurrent Events
The guideline provides rigorous expectations for missing data handling. It states that methods must be conservative under the scenario most likely to falsely show non-inferiority. The guidance warns against using methods that artificially pull treatment arms closer together, especially in equivalence trials.Intercurrent events must be prespecified and handled according to the estimand, with special attention to events that can mask true differences.
9. Multiple Objectives and Prohibitions on Post-Hoc Switching
The guideline states that sponsors may include multiple predefined objectives, but these must be statistically controlled via multiplicity procedures.However, the guidance strongly warns against changing a superiority trial into a non-inferiority trial after initiation, stating that such changes undermine credibility, violate pre-specification requirements, and risk biased outcomes.
10. Annex: How Historical Evidence Should Be Assessed
The annex of the guidance explains how sponsors should select, review, and justify historical comparator studies. It details risks like selection bias, publication bias, outdated standards of care, and changing disease characteristics. It emphasises that historical trials must be relevant to the current trial’s population, design, and endpoint definitions.
Sponsors should carefully incorporate these principles to ensure regulatory acceptance and high-quality trial outcomes.



Comments